
About ALS (Amyotrophic Lateral Sclerosis)
Physical Health

Amyotrophic lateral sclerosis (ALS) — previously referred to as Lou Gehrig’s disease —is a fatal neurological disorder that affects the brain and spinal cord. ALS is a progressive disease, meaning that once the symptoms begin, a person’s condition continues to worsen over time. Unfortunately, there’s currently no cure for ALS. However, doctors and researchers have discovered several medications that help slow disease progression, giving people with ALS more time with fewer symptoms.
This article will provide an overview of ALS and what you need to know — including information about symptoms, diagnosis, and the latest treatments.
What Is ALS?
Your brain and spinal cord are made of billions of nerve cells (known as neurons) that work together to send signals to one another. Motor neurons are a specific type of neuron that sends signals from the brain and spinal cord to the muscles, telling them to move. There are 2 types of motor neurons:
- Upper motor neurons: Located in the brain and spinal cord
- Lower motor neurons: Located in the brain stem (at the base of the brain) and the spinal cord
When you want to move your arm or leg, your brain communicates with the upper motor neurons, which send signals to the lower motor neurons. From there, signals are sent out from the spinal cord to the periphery nerves located in the muscles. In ALS, the motor neurons die off and can no longer send signals to the muscles. Without enough functioning nerve cells, everyone with ALS eventually loses the ability to control their muscles and movements.
What Causes ALS?
Doctors and researchers still aren’t sure what causes ALS. They believe it may be due to a combination of genetics and environmental risk factors.
Nearly all (90%) of ALS cases occur randomly, meaning that a person has no obvious risk factors causing their disease. On the other hand, 10% of people with ALS have a family history of the disease. This means they have a parent or sibling who also has ALS. Doctors and researchers refer to it as familial ALS because some families have certain gene changes (mutations) passed down. According to the Mayo Clinic, parents with familial ALS have a 50% chance of passing their disease on to their children.
Up to 40% of familial ALS cases are caused by changes in the C9orf72 gene. This gene makes a specialized protein found in motor neurons in the brain. The National Institute of Neurological Disorders and Stroke (NINDS) states that another 12-20% of familial ALS cases are caused by changes in the SOD1 gene. This gene creates an enzyme known as superoxide dismutase, which destroys superoxide radicals in the body. Mutations in the SOD1 gene mean there are more toxic radicals damaging healthy cells throughout the body.
Risk Factors for ALS
Certain factors can raise your risk of developing ALS. Some of these factors are within your control, while others aren’t. If you’d like to learn more about your individual risk of ALS, talk with your doctor or healthcare provider.
One large review of 163 studies found that the following factors increase the risk of ALS:
- Exposure to heavy metals (such as lead, arsenic, or mercury) or pesticides (insecticides and herbicides)
- Having a previous head injury
- Military service
- Exposure to electromagnetic fields, especially in those working as welders
- Stroke
- High blood pressure (hypertension)
Other studies have also found that smoking significantly increases the risk of ALS — by up to 70%. Exposure to the toxic chemicals in cigarette smoke may be to blame. The Mayo Clinic notes that postmenopausal women (those who have undergone menopause) who smoke are at an even higher risk of ALS.
Your age and sex can also play a role in your chances of developing ALS. The average age of an ALS diagnosis is 55 years old — but most people are diagnosed between the ages of 40 and 70. Younger adults can develop ALS as well, but it’s less common. Men are 20% more likely to develop ALS compared to women. This is especially true for men under the age of 65. After the age of 70, men and women are equally as likely to develop the disease.
Symptoms of ALS
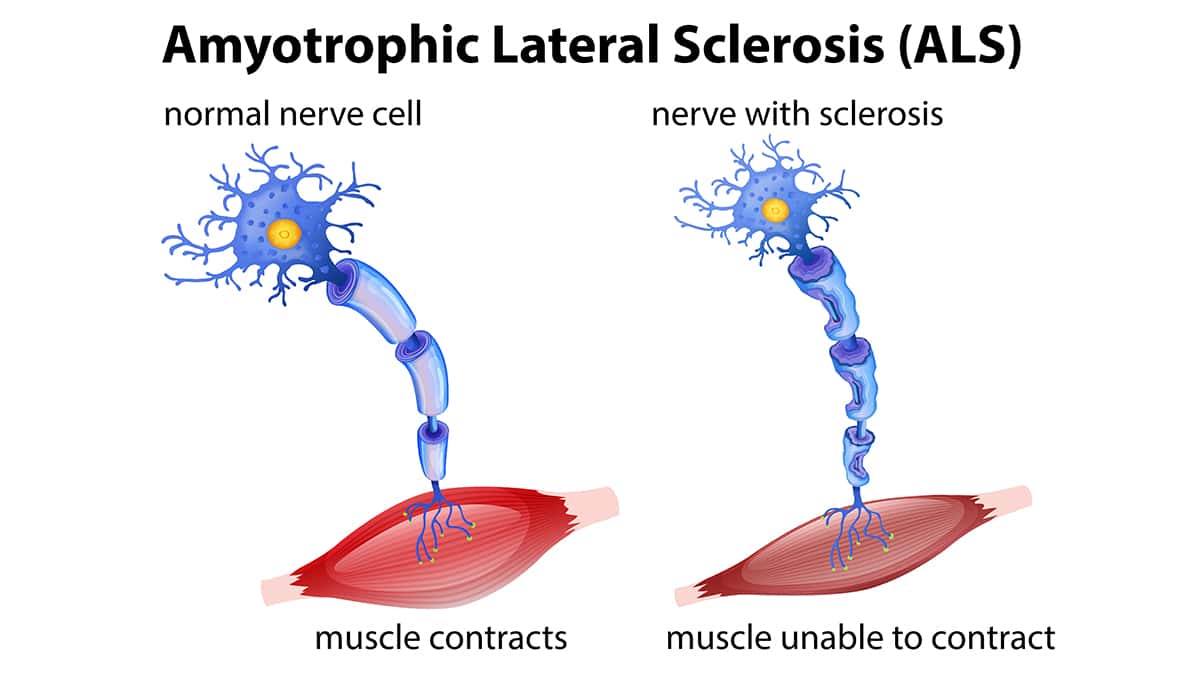
Your ALS symptoms depend on which motor neurons are affected. Most people with ALS have both upper and lower motor neuron dysfunction. As these nerve cells start to die off, you’ll start noticing some signs of ALS. Upper motor neuron degeneration typically causes poor balance, loss of coordination, and muscle tightness (spasticity). Lower motor neuron degeneration can cause muscle twitching, weakness, and shrinkage (atrophy).
ALS symptoms may start in one area of the body but can quickly spread to others. The early signs of ALS can include:
- Muscle cramping and twitching in the tongue, shoulders, arms, hands, or legs
- Muscle spasms
- Trouble chewing and swallowing
- Muscle weakness in the neck, arms, or legs
- Thick, slurred, or nasal speech that makes it difficult to project your voice
- Fatigue or extreme tiredness
- Trouble chewing and swallowing
As ALS progresses, your brain continues losing the ability to send signals to motor neurons. This means you may not be able to move your muscles on your own. If your ALS has progressed to later stages, you may have trouble:
- Breathing, leading to shortness of breath
- Chewing and swallowing food, known as dysphagia
- Maintaining a healthy weight and getting the nutrients your body needs
- Urinating and having bowel movements
- Forming words and speaking, known as dysarthria
- Controlling outbursts of unintended laughing or crying, known as the pseudobulbar affect
- With drooling, known as sialorrhea
Eventually, everyone with ALS loses their ability to stand up, walk, and eat on their own. Most people die because they can’t breathe on their own (known as respiratory failure). After receiving an ALS diagnosis, the typical lifespan is 3-5 years. However, NINDS notes that around 10% of people with ALS live for a decade or more after their diagnosis.
Diagnosing ALS
There is currently no single test available to diagnose ALS. Instead, your doctor will use the results of your physical exam, medical history, nerve and muscle tests, and imaging scans to make a final diagnosis.
Physical and Neurological Exams
During your physical exam, your doctor will ask detailed questions about your medical history. Be sure to tell them about any symptoms you’re experiencing and when they started. If you have a family history of ALS or another neurodegenerative disease, let your doctor know as well. They’ll look at your arms and legs for any signs of twitching, spasticity, or atrophy. Your doctor will also perform a neurological exam to test your muscle strength, reflexes, and sensitivity to touch. During this exam, you may be asked to:
- Move muscles in your tongue, face, arms, and legs
- Walk in a straight line
- Write your name
- Speak
Nerve and Muscle Tests
The next step is to perform electromyography (EMG), which tests how well your nerves and muscles work. There are 2 types of EMG — a needle exam and a nerve conduction study (NCS). Your doctor will likely perform both of these tests at the same time.
As the name suggests, a needle exam uses a needle electrode inserted under your skin and into the muscle fibers. The electrode sends small electric shocks through your nerves to measure how fast they travel. Some people report that this feels similar to a static shock, which can be uncomfortable at times. Your doctor will use this test to rule out another condition known as multifocal motor neuropathy, which can look similar to ALS. A needle exam can also “listen” to how electric signals move through your muscles — this part of the exam doesn’t require any electric shocks.
An NSC measures signals sent from your nerves to your muscles using sticky electrode patches placed on your skin. One electrode sends very small shocks through your nerves, while the other measures the electrical activity. You may notice some tingling during an NCS, but it likely won’t hurt.
Imaging Tests
Your doctor may also use imaging tests like magnetic resonance imaging (MRI) scans to take a closer look at your brain and spinal cord. MRI uses strong magnets and radio waves to create extremely detailed pictures of the body’s soft tissues. An MRI can’t specifically diagnose ALS. Instead, it’s used to rule out other conditions that may be causing your symptoms, including:
- Multiple sclerosis (MS), an autoimmune condition that also affects the brain and spinal cord
- A spinal cord tumor
- A slipped or herniated disc, which occurs when a soft cushion (disc) that sits between the vertebrae (bones) in the spine pushes out of a tear in the tough outer exterior
Other Tests for Diagnosing ALS
If your doctor needs more information to make an ALS diagnosis, they may also order:
- Blood tests to check for thyroid issues, low vitamin levels, autoimmune disorders, or cancer
- Genetic testing to check for genes in familial ALS
- A spinal tap (lumbar puncture) to check your spinal fluid for abnormalities
- A muscle biopsy to check for other neuromuscular conditions
Treatments for ALS
Many people remember the viral “Ice Bucket Challenge” from 2014 when participants dumped buckets of ice water on their heads to raise money for ALS research. Nearly a decade later, doctors and researchers have successfully developed new treatments aimed at reducing ALS symptoms and slowing disease progression. The U.S. Food & Drug Administration (FDA) has approved 4 treatments for ALS.
Riluzole (Rilutek, Tiglutik, ExservanTM)
Riluzole (Rilutek) was approved by the FDA in 1995 for treating ALS. It’s now sold under the generic name riluzole. It works by blocking the release of glutamate — an amino acid that can damage nerve cells in large amounts. Riluzole is taken as a tablet once daily. Studies show that it’s important to start taking this medication as soon as possible. Doctors originally thought riluzole only extended a person’s lifespan by 3 months, but newer studies show it can help people with ALS live up to a year longer.
If you have trouble swallowing due to ALS, your doctor may prescribe you another form of riluzole. Tiglutik is a thickened liquid formulation that’s easier to swallow than a pill. Exservan is an oral film that’s placed on the tongue and quickly dissolves.
Edaravone (RadicavaTM)
Radicava was first approved by the FDA in 2017 for treating ALS. Doctors and researchers aren’t quite sure why Radicava works, but they think it may destroy toxic radicals that damage healthy cells. Radicava was originally formulated as an intravenous (IV) injection, meaning it was injected into a vein in the arm. In 2022, the FDA also approved a liquid formulation taken by mouth or through a feeding tube.
Since Radicava is a relatively new drug, we don’t know how it affects the lifespan of those with ALS yet.
Sodium Phenylbutyrate-Taurursodiol (Relyvrio®)
Relyvrio is a new ALS treatment approved by the FDA in 2022. It helps stop motor neurons from dying by targeting the mitochondria and endoplasmic reticulum (ER) in the cells. Both play an important role in keeping motor neurons healthy and functioning properly.
Clinical studies show that Relyvrio slows disease progression in people with ALS by 25%. This medication may also help them live 6 months longer than those who don’t take it.
Relyvrio comes as a powder that you mix into a glass of room-temperature water. You may notice the medication has an unpleasant taste.
Tofersen (Qalsody®)
If you have familial ALS with an SOD1 mutation, your doctor will likely prescribe you Qalsody. Approved by the FDA in 2023, Qalsody prevents cells from making mutated SOD1 proteins. Clinical studies showed that treatment helped prevent nerve injury and neurodegeneration in people with ALS and SOD1 mutations.
Qalsody is given as an injection in the spine (intrathecal injection) every 14 days for 3 initial doses. After that, you’ll receive a maintenance injection every 28 days. Since Qalsody is a new treatment, we don’t know yet how it affects the lifespan of people with ALS.
Supportive Treatments
If you have ALS, you may need additional supportive treatments to help you live more comfortably. The Mayo Clinic gives several examples, including:
- Breathing support, including a ventilator mask or a tracheostomy (a surgical procedure that creates a hole in the windpipe for a ventilator)
- Physical, speech, and occupational therapy
- Nutritional support from a registered dietitian
- Emotional and psychological support from therapists and social workers


